Abstract
Background
AKT pathway activation is implicated in endocrine-therapy resistance. Data on the efficacy and safety of the AKT inhibitor capivasertib, as an addition to fulvestrant therapy, in patients with hormone receptor–positive advanced breast cancer are limited.
Methods
In a phase 3, randomized, double-blind trial, we enrolled eligible pre-, peri-, and postmenopausal women and men with hormone receptor–positive, human epidermal growth factor receptor 2–negative advanced breast cancer who had had a relapse or disease progression during or after treatment with an aromatase inhibitor, with or without previous cyclin-dependent kinase 4 and 6 (CDK4/6) inhibitor therapy. Patients were randomly assigned in a 1:1 ratio to receive capivasertib plus fulvestrant or placebo plus fulvestrant. The dual primary end point was investigator-assessed progression-free survival assessed both in the overall population and among patients with AKT pathway–altered (PIK3CA, AKT1, or PTEN) tumors. Safety was assessed.
Results
Overall, 708 patients underwent randomization; 289 patients (40.8%) had AKT pathway alterations, and 489 (69.1%) had received a CDK4/6 inhibitor previously for advanced breast cancer. In the overall population, the median progression-free survival was 7.2 months in the capivasertib–fulvestrant group, as compared with 3.6 months in the placebo–fulvestrant group (hazard ratio for progression or death, 0.60; 95% confidence interval [CI], 0.51 to 0.71; P<0.001). In the AKT pathway–altered population, the median progression-free survival was 7.3 months in the capivasertib–fulvestrant group, as compared with 3.1 months in the placebo–fulvestrant group (hazard ratio, 0.50; 95% CI, 0.38 to 0.65; P<0.001). The most frequent adverse events of grade 3 or higher in patients receiving capivasertib–fulvestrant were rash (in 12.1% of patients, vs. in 0.3% of those receiving placebo–fulvestrant) and diarrhea (in 9.3% vs. 0.3%). Adverse events leading to discontinuation were reported in 13.0% of the patients receiving capivasertib and in 2.3% of those receiving placebo.
Conclusions
Capivasertib–fulvestrant therapy resulted in significantly longer progression-free survival than treatment with fulvestrant alone among patients with hormone receptor–positive advanced breast cancer whose disease had progressed during or after previous aromatase inhibitor therapy with or without a CDK4/6 inhibitor. (Funded by AstraZeneca and the National Cancer Institute; CAPItello-291 ClinicalTrials.gov number, NCT04305496. opens in new tab.)
Approximately 70% of advanced breast cancers express the hormone receptor estrogen or progesterone (or both) and do not have human epidermal growth factor receptor 2 (HER2) overexpression.1 In these patients, endocrine therapy, often an aromatase inhibitor, combined with a cyclin-dependent kinase 4 and 6 (CDK4/6) inhibitor is the mainstay of first-line treatment for advanced disease. Such treatment has been shown to improve progression-free and overall survival substantially as compared with aromatase inhibitor therapy alone.2,3 Nevertheless, most patients have disease progression,4 and treatment of these patients remains a clinical challenge.2 Appropriate endocrine-based treatment after disease progression during aromatase inhibitor therapy, with or without a CDK4/6 inhibitor, is unclear. However, options include the selective estrogen-receptor degrader fulvestrant as monotherapy3 or as part of combination treatment.2,3
AKT is the key node of the phosphatidylinositol 3-kinase (PI3K)–AKT–PTEN signaling pathway. Overactivation of the pathway occurs in approximately half of hormone receptor–positive, HER2-negative breast cancers by means of activating mutations in PIK3CA and AKT1 and inactivating alterations in PTEN.5-7 Alterations may be present at the time of cancer recurrence and can also be acquired by means of previous treatment, including with CDK4/6 inhibitors.8,9 AKT signaling may also be activated in the absence of genetic alterations in patients with endocrine resistance.10-12 Inhibition of this pathway has been successful in pretreated hormone receptor–positive advanced breast cancer — results that led to regulatory approval.13-15 The PI3K α-selective inhibitor alpelisib, combined with fulvestrant, in PIK3CA-mutant tumors in the SOLAR-1 (Clinical Studies of Alpelisib in Breast Cancer 1) trial13 and the mammalian target of rapamycin (mTOR) inhibitor everolimus, combined with exemestane, in the BOLERO-2 (Breast Cancer Trials of Oral Everolimus-2) trial had greater efficacy than endocrine therapy alone.14,15 Both randomized trials were conducted before the availability of CDK4/6 inhibitors, but in a follow-on phase 2, single-group trial (BYLieve), investigators attempted to address the need for data in this context with alpelisib plus fulvestrant.16
Capivasertib is an orally bioavailable, small-molecule inhibitor of all three AKT isoforms.17 Capivasertib inhibited AKT in preclinical models, resulting in dephosphorylation of key downstream targets; the drug also showed antiproliferative activity in breast-cancer cell lines and had synergistic antitumor activity in combination with endocrine therapy in preclinical models.17,18 In the phase 2 FAKTION trial, treatment with capivasertib in combination with fulvestrant significantly improved progression-free and overall survival as compared with fulvestrant alone among postmenopausal women with hormone receptor–positive advanced breast cancer who had previously received endocrine therapy.19,20 Here, we present the primary analysis of CAPItello-291, a phase 3 trial that assessed the efficacy and safety of capivasertib–fulvestrant therapy in patients with hormone receptor–positive, HER2-negative advanced breast cancer whose disease had progressed during or after aromatase inhibitor therapy, with or without a CDK4/6 inhibitor.
Methods
Trial Design
In this randomized, double-blind, placebo-controlled, phase 3 trial, we randomly assigned patients in a 1:1 ratio to receive either oral capivasertib (at a dose of 400 mg twice daily for 4 days, followed by 3 days off) plus fulvestrant (500 mg, administered intramuscularly every 14 days for the first three injections and every 28 days thereafter) or matching placebo plus fulvestrant. One cycle was defined as 4 weeks of capivasertib or placebo receipt. Premenopausal or perimenopausal women also received a luteinizing hormone–releasing hormone agonist for the duration of the trial treatment period. Randomization was stratified according to the presence or absence of liver metastases, previous use of a CDK4/6 inhibitor (yes or no), and geographic region.
Treatment continued until disease progression (assessed according to the Response Evaluation Criteria in Solid Tumors [RECIST], version 1.1), the occurrence of unacceptable toxic effects, withdrawal of consent, or death. Dose reduction of capivasertib or placebo was allowed from 400 mg twice daily to 320 mg twice daily, and then to 200 mg twice daily, if indicated. Dose reductions or interruptions were made for the occurrence of adverse events of grade 3 or higher that were attributed to capivasertib or placebo or for lower grades of adverse events as clinically appropriate. Reductions in the dose of fulvestrant were not allowed. Patients who discontinued capivasertib or fulvestrant for reasons other than disease progression could continue to receive the other. No primary prophylaxis was permitted for rash and diarrhea, but investigators were advised to consider secondary prophylaxis in specific circumstances (see the protocol, available with the full text of this article at NEJM.org).
Patients
Premenopausal, perimenopausal, or postmenopausal women or men (≥18 years of age in most regions; ≥20 years in Japan) with hormone receptor–positive, HER2-negative locally advanced (i.e., primary inoperable) or metastatic breast cancer were eligible. Hormone receptor–positive status was defined as estrogen-receptor expression with or without progesterone-receptor expression, which, along with HER2 status, was assessed locally. HER2-negative status was defined as 0 or 1+ intensity on immunohistochemical testing, as 2+ intensity on immunohistochemical testing and no amplification on in situ hybridization, or if immunohistochemical testing was not done, as no evidence of amplification on in situ hybridization, according to the American Society of Clinical Oncology and College of American Pathologists guideline recommendations.21,22 Patients must have had disease progression while they had previously been receiving an aromatase inhibitor, with or without a CDK4/6 inhibitor; disease progression was defined as progression during previous therapy with an aromatase inhibitor in the context of metastatic disease or as progression during treatment or within 12 months after the end of treatment with a neoadjuvant or adjuvant aromatase inhibitor. Aromatase inhibitor therapy was not required to be the most recent treatment.
Patients were allowed to have received up to two previous lines of endocrine therapy and one previous line of chemotherapy in the context of advanced disease. The trial protocol required the enrollment of a minimum of 51% of patients with previous CDK4/6 inhibitor treatment. Disease progression during previous therapy was required. Patients with previous exposure to fulvestrant or another selective estrogen-receptor degrader or to AKT, PI3K, or mTOR inhibitors were excluded, as were patients with diabetes who were receiving insulin or had a baseline glycated hemoglobin level of at least 8.0% (63.9 mmol per mole).
Patients were required to have measurable disease (assessed according to RECIST, version 1.1) or at least one lytic or mixed lytic–blastic bone lesion with identifiable soft-tissue components that could be assessed by means of computed tomography (CT) or magnetic resonance imaging (MRI). Patients had an Eastern Cooperative Oncology Group performance-status score of 0 or 1 (on a scale from 0 [no disability] to 5 [death]) with no deterioration over the preceding 2 weeks. Tumor tissue for molecular analysis was required. Activating mutations in PIK3CA and AKT1 and inactivating alterations in PTEN genes were determined centrally (after randomization) by means of next-generation sequencing with the use of the FoundationOneCDx assay (Foundation Medicine) in all countries except China (OncoScreen Plus, Burning Rock Biotech). Patients whose tumors had at least one qualifying alteration in these three genes were included in the AKT pathway–altered population. Patients with tumors that did not have a qualifying alteration detected in any of these three genes or with an unknown test result were included in the AKT pathway–nonaltered population.
End Points
The dual primary end point was investigator-assessed progression-free survival (assessed according to RECIST, version 1.1) in the overall population and among patients with AKT pathway–altered tumors. Secondary end points included overall survival, objective response, and safety. Patient-reported end points included the European Organization for Research and Treatment of Cancer (EORTC) Core Quality of Life Questionnaire (QLQ-C30). The QLQ-C30 is assessed on a scale of 0 to 100, with higher scores indicating a higher quality of life.
Procedures
Tumor assessments according to RECIST, version 1.1, were performed with the use of CT or MRI scans (or both) at screening (within 4 weeks before randomization), every 8 weeks for the first 18 months, and then every 12 weeks until disease progression. Radiographic bone scans were performed at screening and repeated as clinically indicated. Patients who discontinued capivasertib or fulvestrant for reasons other than disease progression continued to undergo scans every 8 weeks until disease progression (assessed according to RECIST, version 1.1).
Biochemical and hematologic tests and vital signs were assessed on days 1 and 15 of the first two cycles and on day 1 of subsequent cycles. Fasting glucose levels were assessed on days 1 and 15 of the first cycle and then every 4 weeks until the discontinuation of capivasertib or placebo. Adverse effects were recorded continuously until 30 days after the discontinuation of capivasertib, fulvestrant, or placebo and were graded according to the National Cancer Institute Common Terminology Criteria for Adverse Events (CTCAE), version 5.0.
Trial Oversight
The trial was designed and overseen by a steering group of medical oncology experts, including representatives from AstraZeneca (the sponsor). The investigators gathered the data. Data analyses were performed by a clinical research organization (Covance) and paid for by the sponsor. The authors vouch for the completeness and accuracy of the data and for the fidelity of the trial to the protocol. An institutional review board and an independent ethics committee reviewed the trial protocol, amendments, and other relevant documents. All the patients provided written informed consent before enrollment.
The trial was conducted in accordance with the applicable International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use and Good Clinical Practice guidelines and with the principles of the Declaration of Helsinki. An independent data and safety monitoring committee assessed the progress of the trial approximately every 6 months and reviewed unblinded safety data. The first draft of the manuscript was prepared by the first author in collaboration with authors employed by the sponsor. Editorial and medical writing assistance with subsequent drafts was provided by paid representatives of the sponsor who are not authors. Subsequently, all the authors were involved with interpreting the data and with writing and reviewing the manuscript.
Statistical Analysis
This trial was powered to assess the effect of capivasertib therapy on progression-free survival and overall survival. A total sample of 700 patients was planned. The primary end point was to be analyzed at approximately 77% maturity in the overall population (when 542 events of progression or death had occurred) and in the AKT pathway–altered population (when 217 events had occurred), under an assumption that 40% of the trial population would have AKT pathway–altered tumors. Assuming a true hazard ratio of 0.64 for progression or death in the analysis of progression-free survival in both populations, we estimated that the trial would have more than 99% power to show a significant difference in favor of the capivasertib–fulvestrant group in the overall population (at a two-sided P<0.035) and 91% power to show a significant difference in favor of the capivasertib–fulvestrant group in the AKT pathway–altered population (at a two-sided P<0.05), with recycling of the remaining 1.5% alpha.
Overall survival assessments of no detriment (i.e., with the hazard ratio not favoring the placebo–fulvestrant group) in the overall and AKT pathway–altered populations were conducted at the time of the primary analysis of progression-free survival (with the assignment of a 0.01% alpha penalty). Efficacy analyses included all the patients who had undergone randomization. The two primary end points were tested with the use of a log-rank test, with stratification according to the presence or absence of liver metastases, previous use of a CDK4/6 inhibitor (yes or no), and geographic area (assessed in the overall population only). Hazard ratios and associated 95% confidence intervals were calculated from a stratified Cox proportional-hazards model fitted with the use of the PROC PHREG procedure in SAS, version 9.4, software (SAS Institute) with the Efron method to control for ties. Subgroup analyses were performed according to various factors (e.g., AKT pathway–altered status and previous use of a CDK4/6 inhibitor) and are presented as forest plots. The percentage of patients with an objective response was analyzed with the use of a logistic-regression model with adjustment for the randomization stratification factors in both populations.
Safety analyses included all the patients who received at least one dose of capivasertib, fulvestrant, or placebo. Safety data were summarized with the use of descriptive statistics. The statistical analysis plan is available with the trial protocol.
Results
Patients
Between June 2, 2020, and October 13, 2021, a total of 901 patients were enrolled at 193 centers in 19 countries; 708 patients (from 181 sites) underwent randomization, with 355 patients assigned to the capivasertib–fulvestrant group and 353 to the placebo–fulvestrant group (Fig. S1 in the Supplementary Appendix, available at NEJM.org). A total of 289 patients (40.8%) in the overall population had tumors with AKT pathway alterations. In an analysis that excluded the 106 patients (15.0%) with unknown alteration status (no sample available or sample did not meet the specified quality metric), 289 (48.0%) of the 602 patients with tumor-sequencing results had AKT pathway alterations. A total of 313 patients (44.2%) had confirmed AKT pathway–nonaltered tumors (Table S1).
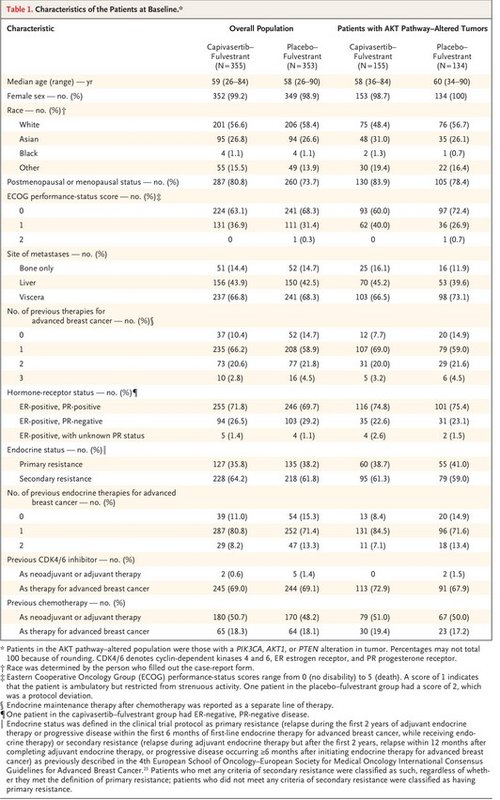
The baseline characteristics of the patients were broadly well-balanced between the two trial groups in both populations (Table 1).23 The median age of the patients was 58 years (range, 26 to 90), and 77.3% of the patients were postmenopausal women. All the patients had HER2-negative disease, 69.1% had previously received a CDK4/6 inhibitor, and 18.2% had previously received chemotherapy for advanced cancer. The demographic characteristics of the patients were largely representative of patients with hormone receptor–positive, HER2-negative breast cancer (Table S2).
Treatment
At the primary analysis (data-cutoff date, August 15, 2022), 63 patients (17.7%) were continuing to receive treatment with capivasertib and 43 (12.3%) were continuing to receive placebo. The median duration of treatment with capivasertib was 5.4 months. Patients in the placebo–fulvestrant group received placebo for a median of 3.6 months. The median duration of treatment with fulvestrant was 5.8 months in the capivasertib–fulvestrant group and 3.7 months in the placebo–fulvestrant group.
Capivasertib was discontinued in 292 patients (82.3%), and placebo was discontinued in 307 (87.7%). The main reason for discontinuation of capivasertib or placebo was disease progression, which occurred in 209 patients (58.9%) receiving capivasertib–fulvestrant and in 273 (78.0%) receiving placebo–fulvestrant.
Efficacy
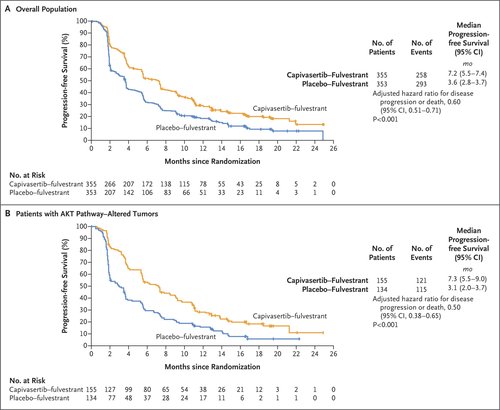
Figure 1. Investigator-Assessed Progression-free Survival in the Overall Population and in Patients with AKT Pathway–Altered Tumors.
The primary analysis was conducted after the occurrence of 551 events of disease progression or death (258 events in the capivasertib–fulvestrant group and 293 in the placebo–fulvestrant group). In the overall population, the median progression-free survival according to the investigator’s assessment was 7.2 months in the capivasertib–fulvestrant group, as compared with 3.6 months in the placebo–fulvestrant group (hazard ratio for progression or death, 0.60; 95% confidence interval [CI], 0.51 to 0.71; P<0.001) (Figure 1A). In the AKT pathway–altered population, in which 236 events of progression or death had occurred (121 events in the capivasertib–fulvestrant group and 115 in the placebo–fulvestrant group), the median progression-free survival was 7.3 months in the capivasertib–fulvestrant group, as compared with 3.1 months in the placebo–fulvestrant group (hazard ratio, 0.50; 95% CI, 0.38 to 0.65; P<0.001) (Figure 1B).
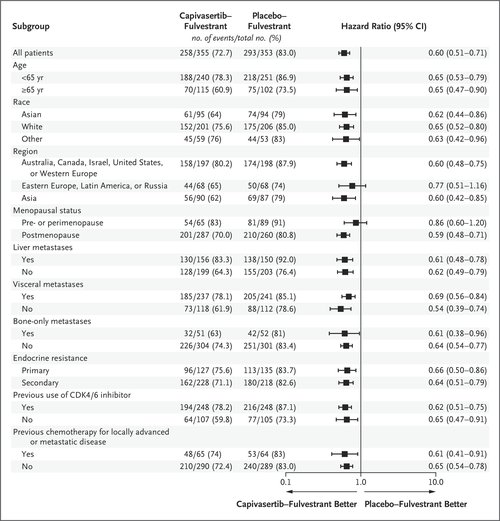
Exploratory analyses of progression-free survival are shown in Figure S2 for patients with AKT pathway–nonaltered tumors, including those with an unknown result on next-generation sequencing (419 patients; hazard ratio for progression or death in per-protocol analysis, 0.70; 95% CI, 0.56 to 0.88); for patients with AKT pathway–nonaltered tumors, excluding patients with unknown results on next-generation sequencing (313 patients; hazard ratio, 0.79; 95% CI, 0.61 to 1.02); and for patients with an unknown result on next-generation sequencing (106 patients; hazard ratio in post hoc analysis, 0.52; 95% CI, 0.32 to 0.83). Subgroup analyses of progression-free survival are shown in Figure 2 and Figure S3. Progression-free survival among patients with and those without previous CDK4/6 inhibitor exposure is shown in Figure S4. Progression-free survival was also assessed on the basis of blinded independent central review; the results were highly consistent with the investigator-assessed analyses, both in the overall population (hazard ratio, 0.61; 95% CI, 0.50 to 0.73) and in the AKT pathway–altered population (hazard ratio, 0.51; 95% CI, 0.38 to 0.68) (Fig. S5). The percentages of patients with an objective response are shown in Table S3.
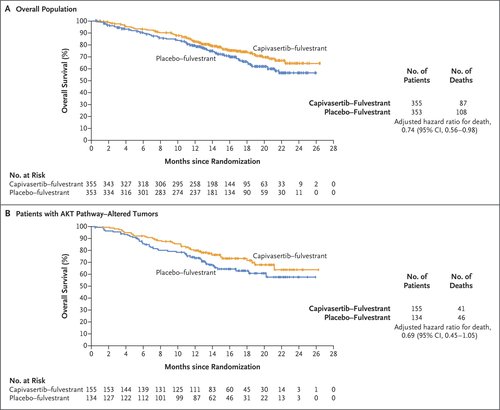
At the time of analysis, 195 patients (27.5%) had died (87 in the capivasertib–fulvestrant group and 108 in the placebo-fulvestrant group). The estimated overall survival at 18 months was 73.9% (95% CI, 68.3 to 78.7) in the capivasertib–fulvestrant group and 65.0% (95% CI, 58.7 to 70.6) in the placebo–fulvestrant group in the overall population (hazard ratio for death, 0.74; 95% CI, 0.56 to 0.98) and 73.2% (95% CI, 64.8 to 80.0) in the capivasertib–fulvestrant group and 62.9% (95% CI, 53.1 to 71.2) in the placebo–fulvestrant group in the AKT pathway–altered population (hazard ratio, 0.69; 95% CI, 0.45 to 1.05) (Figure 3).
Quality of Life
Global health status and quality of life were maintained in both the capivasertib–fulvestrant group and the placebo-fulvestrant group (mean overall change from baseline in the QLQ-C30 score at visits during the treatment period, −2.52 points and −5.62 points, respectively; difference, 3.10 points; 95% CI, 0.21 to 5.98). Global health status and quality of life were maintained for longer with capivasertib–fulvestrant than with placebo-fulvestrant. The median time to deterioration (defined as a sustained decrease of ≥10 points in the score from baseline) was 24.9 months in the capivasertib–fulvestrant group, as compared with 12.0 months in the placebo-fulvestrant group (hazard ratio, 0.70; 95% CI, 0.53 to 0.92). The results in the AKT pathway–altered population were similar to those in the overall population.
Safety
Table 2. Most Frequent Adverse Events in the Overall Population (Safety Population).
The safety population included 355 patients who received capivasertib–fulvestrant and 350 who received placebo–fulvestrant. The most common adverse events of any grade that were reported in the capivasertib–fulvestrant group were diarrhea (in 72.4% of the patients, vs. 20.0% of those in the placebo–fulvestrant group), rash (as a grouped term; in 38.0% and 7.1%, respectively), and nausea (in 34.6% and 15.4%) (Table 2). Hyperglycemia of any grade occurred in 16.3% of the patients who received capivasertib–fulvestrant and in 3.7% of those who received placebo–fulvestrant. The most frequently reported adverse events of grade 3 or higher were rash (as a grouped term; in 12.1% of the patients in the capivasertib–fulvestrant group and in 0.3% of those in the placebo–fulvestrant group), diarrhea (in 9.3% and 0.3%, respectively), and hyperglycemia (in 2.3% and 0.3%). The safety profile of capivasertib–fulvestrant in the AKT pathway–altered population was similar to that in the overall population.
Serious adverse events occurred in 57 patients (16.1%) receiving capivasertib–fulvestrant and in 28 (8.0%) receiving placebo–fulvestrant (Table S4). Death due to adverse events occurred in 4 patients (1.1%) receiving capivasertib–fulvestrant (from acute myocardial infarction, cerebral hemorrhage, aspiration pneumonia, and sepsis in 1 patient each) and in 1 patient receiving placebo–fulvestrant (from coronavirus disease 2019). None of the deaths were considered by the local investigators to be related to capivasertib or fulvestrant.
Adverse events leading to a dose interruption occurred in 124 patients (34.9%) receiving capivasertib, as compared with 36 (10.3%) receiving placebo. Adverse events leading to dose reduction occurred in 70 patients (19.7%) receiving capivasertib, as compared with 6 (1.7%) receiving placebo. Discontinuation due to adverse events occurred in 46 patients (13.0%) receiving capivasertib and in 8 (2.3%) receiving placebo.
Discussion
This double-blind, phase 3, randomized trial, CAPItello-291, showed that the addition of capivasertib to fulvestrant therapy resulted in a significant improvement in progression-free survival among patients with hormone receptor–positive, HER2-negative advanced breast cancer that had progressed during previous aromatase inhibitor therapy with or without a CDK4/6 inhibitor. The safety profile showed that diarrhea and rash were the most common adverse events, and the incidence of discontinuation due to adverse events was relatively low.
Exploratory analyses in the AKT pathway–nonaltered population showed that capivasertib has activity beyond AKT pathway–altered tumors. These results are in line with preclinical data showing that capivasertib inhibited growth in some cell lines without AKT pathway gene alterations,17 a finding that reflects the critical role of the AKT pathway in cancer biology, whereby aberrant signaling can also be increased by other mechanisms.24,25 For example, in untreated triple-negative breast cancer, AKT activation in the absence of AKT pathway alterations is associated with other mechanisms of pathway activation, including loss of PTEN protein expression and enhanced activation of upstream receptor tyrosine kinases.26 Moreover, cross-talk between the AKT and estrogen-receptor pathway may limit the activity of monotherapy, which suggests that simultaneous inhibition of these two pathways could enhance efficacy independent of mutation status.11,18,27 It should be noted that AKT pathway alterations acquired by means of previous therapy may not have been present in the tissue analyzed in this trial.8,9
Our findings suggest that capivasertib–fulvestrant treatment improved outcomes regardless of previous exposure to a CDK4/6 inhibitor and reinforce the evidence that single-agent endocrine therapy has poor outcomes after receipt of a CDK4/6 inhibitor. Among patients who received placebo–fulvestrant, of whom 69% had received a CDK4/6 inhibitor previously for advanced breast cancer, the median progression-free survival was 3.6 months. In the control group of the EMERALD trial, in which all the patients were required to have received a CDK4/6 inhibitor previously, the median progression-free survival with endocrine therapy was 1.9 months.28 In other similar trials before the widespread use of CDK4/6 inhibitors, the median progression-free survival in placebo–fulvestrant groups was 4.0 to 5.7 months.13,29,30
In this trial, randomization was not stratified according to AKT pathway alteration. This approach potentially allowed for the inclusion of patients with more aggressive disease who might otherwise not have enrolled in a trial that mandated waiting for tissue-testing results from a central laboratory before randomization. As a result, the population of our trial probably reflects the clinical heterogeneity seen in routine practice.
The safety profile of capivasertib compared favorably with that of other agents targeting the PI3K–AKT–PTEN pathway that are used in this patient population. Stomatitis (according to CTCAE, version 3.0) was a frequently reported adverse event with everolimus in the BOLERO-2 trial (with events of any grade in 59% of the patients and of grade ≥3 in 8%),31 although subsequent implementation of guidelines recommending the use of dexamethasone mouthwash to manage toxic effects has reduced rates of this event.32 Hyperglycemia (according to CTCAE, version 4.03) was prevalent with alpelisib therapy in the SOLAR-1 trial (with events of any grade in 64% of the patients and of grade ≥3 in 37%), which led to a high incidence of discontinuation.13 Although the incidences of adverse events across the BOLERO-2, SOLAR-1, and CAPItello-291 trials are not directly comparable owing to the different CTCAE versions that were used, as well as to inherent differences among the trials, the incidence and grades of stomatitis and hyperglycemia were low with capivasertib. The CAPItello-291 trial implemented an intermittent administration schedule of capivasertib, which was selected early in clinical development and was due to in part to preclinical modeling, in order to maximize AKT inhibition and optimize the therapeutic window. It is possible that the reduced toxic-effect profile of capivasertib, with a low incidence of hyperglycemia, reflects this intermittent schedule.
In the CAPItello-291 trial, we found that the addition of capivasertib to fulvestrant treatment led to a significant improvement, as compared with fulvestrant alone, in progression-free survival among patients with hormone receptor–positive, advanced breast cancer who had disease progression during or after previous endocrine therapy, with or without a CDK4/6 inhibitor. Diarrhea and rash were the main adverse events in patients treated with capivasertib.
Supported by AstraZeneca and in part by a grant (P30-CA008748) to the Memorial Sloan Kettering Cancer Center from the National Cancer Institute.
Disclosure forms provided by the authors are available with the full text of this article at NEJM.org.
A data sharing statement provided by the authors is available with the full text of this article at NEJM.org.
We thank all the patients and their families; all the trial-site coordinators and nurses; the members of the independent data and safety monitoring committee; and Alison Lovibond, Ph.D., and Suzanne Patel, Ph.D., of BOLDSCIENCE, for editorial and medical writing assistance with an earlier version of the manuscript.
Author Affiliations
From the Royal Marsden Hospital, Institute of Cancer Research, London (N.C.T.), the Christie NHS Foundation Trust, Manchester (S.J.H.), and Oncology Research and Development, AstraZeneca, Cambridge (E.C.B., L.G., G.S., A.F.) — all in the United Kingdom; the Department of Medical Oncology, Vall d’Hebron University Hospital (M. Oliveira), the Breast Cancer Unit, Vall d’Hebron Institute of Oncology (M. Oliveira), the Department of Oncology, International Breast Cancer Center, Pangaea Oncology, Quiron Group, Medica Scientia Innovation Research (J.C.), and Institut de Recerca Biomèdica (S.M.M.), Barcelona, and the Department of Medicine, Faculty of Biomedical and Health Sciences, Universidad Europea de Madrid, Madrid (J.C.) — all in Spain; Institut Claudius Regaud, Institut Universitaire du Cancer–Oncopole Toulouse, Toulouse, France (F.D.); Departamento de Oncología Médica, Instituto Nacional de Enfermedades Neoplásicas, and Universidad Ricardo Palma — both in Lima, Peru (H.L.G.M.); Shanghai Cancer Center, Fudan University, Shanghai, China (X.H.); Memorial Sloan Kettering Cancer Center and Weill Cornell Medical College — both in New York (K.J.); Petrov Research Institute of Oncology, St. Petersburg (P.K.), and Loginov Moscow Clinical Scientific Center, Moscow (L.Z.) — both in Russia; GBG Forschungs, Neu-Isenburg, and the Center for Hematology and Oncology, Bethanien, Frankfurt — both in Germany (S.L.); Icon Cancer Centre, Adelaide, SA, Australia (M. Okera); Sungkyunkwan University School of Medicine, Samsung Medical Center (Y.H.P.), and Yonsei University College of Medicine, Yonsei Cancer Center (J.S.) — both in Seoul; Kyoto University Hospital, Kyoto (M.T.), and National Hospital Organization Kyushu Cancer Center, Fukuoka (E.T.) — both in Japan; Emek Medical Center, Afula, Israel (S.Y.); and the University of California San Francisco Helen Diller Family Comprehensive Cancer Center, San Francisco (H.S.R.).
Dr. Turner can be contacted at nick.turner@icr.ac.uk or at the Royal Marsden Hospital, Institute of Cancer Research, Fulham Rd., London SW3 6JJ, United Kingdom.
A list of the members of the CAPItello-291 Study Group is provided in the Supplementary Appendix, available at NEJM.org.

