AL Amyloidosis Background
Every protein has an assigned job, a schedule, and a freshly pressed lab coat. Antibodies clock in, fight infections, and clock out when their job is done. But what happens when one of those antibodies becomes deceitful—cutting corners, breaking the rules, or acting outside its assigned role? That’s when light chain AL amyloidosis can develop. Light chain AL amyloidosis is a rare and often underdiagnosed medical condition, characterized by multiorgan deposition of misfolded immunoglobulin light-chain amyloid fibrils that cause mechanical disruption, local oxidative stress, and eventual organ failure.1 The most involved organs are the heart and kidneys, but the gastrointestinal tract, liver, and nerves may also be involved. The clinical presentation of AL amyloidosis often includes fatigue and unintentional weight loss but may also include additional symptoms specific to the organs involved.2-4
The clinical evaluation of AL amyloidosis begins with routine laboratory tests, including a complete blood count (CBC) with differential and a comprehensive metabolic panel (CMP). Additionally, serum protein electrophoresis with immunofixation will assess for the presence of a monoclonal paraprotein, while urine protein electrophoresis will quantify proteinuria and characterize urinary light chains. Serum free light chains should also be included in the diagnostic workup and, if present, confirmed by tissue biopsy. A fat pad biopsy is a specialized procedure where subcutaneous adipose tissue, typically from the abdomen, is collected for Congo red staining to confirm the presence of amyloid, but not the type of amyloid. The reported sensitivity is approximately 85% with multiple contributing factors that can affect the result: operator expertise, tissue handling, and biopsy site. A bone marrow biopsy and aspiration can confirm amyloid deposition. Once amyloidosis is identified, mass spectrometry-based proteomic analysis will assist in determining the type of amyloid present.1-4
Treatment Strategies for AL Amyloidosis
After a diagnosis is confirmed, the central questions are: how should we treat AL amyloidosis, and what is the likelihood of achieving a complete hematologic response (CHR)? A CHR in AL amyloidosis is defined as negative immunofixation in serum and urine and normalization of the involved serum free light chains (FLC). As a hematological response typically takes about 6-12 months as plasma cell dyscrasia responds to treatment. Organ function improvement on the other hand may take up to 24 months as amyloid deposits need to be reabsorbed for reversal of damage to be seen.5
Before the ANDROMEDA trial, for eligible patients, the treatment paradigm often centered on proceeding to autologous stem cell transplant after high-dose melphalan conditioning to achieve a CHR, with results in studies from the 1990s to early 2000s ranging from 40% to 60%.6 High-dose melphalan followed by ASCT was compared to chemotherapy (melphalan and dexamethasone) by Gertz et al. showing a median progression free survival (PFS) (4 vs. 1.63 years) and median overall survival (not achieved vs. 6.53 years) to favor ASCT, while Jaccard et al did not show superiority between either arm.7,8 The comparison between efficacy and safety is also inconclusive as unexpected early mortality occurred in the ASCT arm and 26% of patients assigned to a transplant did not receive one.9 Treatment-related mortality was noted to be higher in patients with cardiac involvement, renal impairment, or advanced age in AL amyloidosis compared to multiple myeloma due to imminent risks like organ damage from existing amyloid fibrils and mortality related to cardiotoxic conditioning.6,9 Multiple trials have compared a dose reduction of melphalan conditioning from 200 mg/m2 to 100-140 mg/m2. However, variable CHR has been noted and showed a trend of decreased overall survival from approximately 10 years to 5 years with the dose reduction.10 Given that cardiac and renal involvement are present in approximately 75% and 60% of patients, respectively, and that the median age at diagnosis is approximately 65 years, a very small number of patients actually qualify for an ASCT with a good performance status and limited organ involvement, which resulted in an unmet need in the treatment strategies and role for ASCT in AL amyloidosis.3
Treatment has evolved to target the underlying etiology of AL amyloidosis by utilizing multiple myeloma regimens, rarely Non-Hodgkin lymphoma regimens, to achieve organ and light-chain response, reduce clonal expansion, and improve patient tolerance prior to ASCT. Bortezomib-based regimens showed promising results after Kastritis et al. published a phase 3 trial that compared melphalan and dexamethasone (MDex) with bortezomib, melphalan, and dexamethasone (BMDex) in newly diagnosed AL amyloidosis. A total of 78% of patients who received BMDex, compared with 53% (P=0.001) of patients who received MDex, met the primary endpoint of a hematologic response at 3 months. Patients who achieved a cardiac response were still on the lower end and favored the BMDex (38% vs. 24%) along with a lower percentage of cardiac progression (BMDex 32% vs. MDex 15%, p=0.54). Renal response was similar in each arm. Overall, after a median of 25 months, survival did not differ significantly between the two groups.11 Pallladini et al. combined bortezomib with a different alkylator, cyclophosphamide, along with dexamethasone (CyBorD) and showed a hematologic response in 60% of patients, with 23% of those patients achieving a complete response (CR). The trial projected that 55% of patients would survive 5 years, with a median time to second therapy of 13 months.12 Smaller retrospective studies corroborated these results, demonstrating similar hematological responses, with Venner et al. demonstrating an overall hematologic response of 81.4% and an estimated 2-year PFS of 66.5% in 48 patients, while Mikhael et al. demonstrated a 94% overall response and 71% CHR in 16 patients in a smaller cohort.13-14
| Mayo 2012 Staging System | ||||
| Prognostic Risk Variables | Value | Prognostic Variable Risk Score | Stage Based on Prognostic Variable Risk Scores | |
| Troponin | Cardiac troponin T (cTNT) | 0.025 μg/L and higher | 1 | Stage I (Risk Score = 0) |
| High-senstivity cardiac troponin T(hs-cTnT) | 40 pg/mL and higher | Stage II (Risk Score = 1) | ||
| Cardiac troponin I (cTNl) | 0.1 μ/L and higher | |||
| BNP | N-terminal pro- B-type natriuretic peptide (NT-ProBNP) | 1800 ng/L or higher | 1 | Stage III (Risk Score = 2) |
| B-type natriuretic peptide (BNP) | 400 ng/L or higher | |||
| dFLC | Difference between involved and uninvolved serum free light chains (dFLC) | 18 mg/dL or higher (180 mg/L) | 1 | Stage IV (Risk Score = 3) |
1b: Staging for AL Amyloidosis Based on Renal Involvement
| Staging | Palladini et al (2014) | Kastritis et all (2017) | Basset et al (2022) |
| Stage I | Estimated glomerular filtration rate (eGFR) > 50 mL/min and Proteinuria < 5 g/24 h | 24h urinary protein (UPr)/eGFR ratio < 30 | eGFR > 50 mL/min and urine albumin-to-creatinine ratio (UACR) < 3600 mg/g |
| Stage II | eGFR < 50 mL/min or Proteinuria > g/24 h | 24h UPr/eGFR ratio 30-99 | eGFR < 50 mL/min or UACR 3600 mg/g and higher |
| Stage III | eGFR < 50 mL/min and Proteinuria > 5 g/24 h | 24h UPr/eGFR ratio 100 and higher | eGFR < 50 mL/min and UACR 3600 mg/g and higher |
1c: Exclusion Criteria from ANDROMEDA Trial
| Cardiac Exclusion | Renal Exclusion | Other Clinical Exclusion |
|
• eGFR < 20 mL/min/1.73 m² |
|
The ANDROMEDA trial transformed treatment options for newly diagnosed AL amyloidosis patients who remain transplant ineligible. By adding daratumumab, a CD38-targeting monoclonal antibody to CyBorD, the median HCR at 6 months favored quadruplet-based therapy compared to CyBorD alone at 6 months (49.7% vs. 14%). Dara-CyBorD also showed a better cardiac (41.5% vs. 22.2%) and renal response (53% vs. 23.9%). As Dara-CyBorD established superiority as the preferred first-line for newly diagnosed AL amyloidosis, many of the patients with advanced cardiac or renal involvement were excluded: Stage IIIb-IV and estimated glomerular filtration rate less than 20 mL/min/1.73 m2 (see Figure 1c).15 Furthermore, Oubari et al. expanded the understanding of Dara-CyBorD in higher-risk populations. This retrospective study included patients with AL amyloidosis with Mayo stage IIIb and found that patients who received a daratumumab-based regimen reached the median overall survival at the 14.5-month mark of follow-up compared to 6.6 and 2.2 months for the bortezomib-based and other treatment groups, respectively.16 Yohannan et al. also retrospectively evaluated patients who received Dara-CyBorD compared to CyBorD and included advanced cardiac and renal AL amyloidosis. Patients eligible for cardiac response assessment achieved a higher cardiac CR or very good partial response (VGPR) with Dara-CyBorD (46.2% vs. 21.2% [P<0.001] and 76.6% vs. 43.6% [P <0.001]); patients eligible for renal response assessment achieved a higher renal VGPR or better in the Dara-CyBorD group (51.5% vs. 28.8% [P<0.001]).17 These findings indicate that for symptomatic patients with advanced cardiac involvement, Dara-CyBorD remains the best available treatment, provided cardiology clearance is obtained beforehand. Patients with Mayo stage IIIb with poor prognosis may start with monotherapy daratumumab and then re-evaluate the role or Dara-CyBorD.18 Similarly, patients with symptomatic renal AL amyloidosis, regardless of their eGFR, may also receive Dara-CyBorD after further discussion with nephrology.
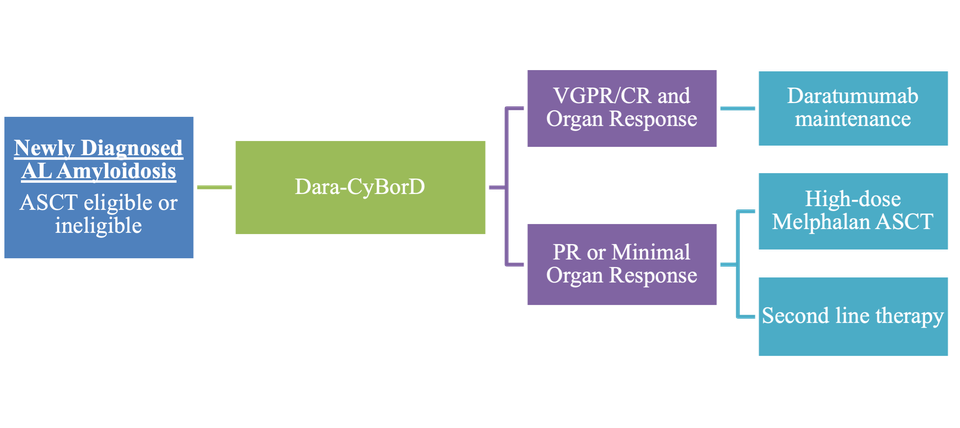
As induction therapy with Dara-CyBorD became the preferred front-line regimen for AL amyloidosis, the question remains regarding the place in therapy for high-dose melphalan ASCT. For patients who achieved a VGPR or better with organ response, a melphalan ASCT may potentially be deferred. If only a partial response (PR) or minimal organ response is achieved with Dara-CyBorD, then a high-dose melphalan ASCT may be considered, depending on cardiac and renal involvement.19 A single-center study led by Romera et al. demonstrated that even patients with cardiac amyloidosis could be safely treated with ASCT after achieving CHR from induction therapy. Cardiac response remained stable post-transplant with an NT-proBNP level of 511.5 pg/mL. Additionally, a median follow-up of 3.6 years revealed prolonged disease-free intervals, with only one death reported.20 It’s crucial that patients with a possible indication for high-dose melphalan ASCT be evaluated at an experienced transplant center as superior outcomes have been noted with a reduction of early mortality.21 If the patient is ineligible for ASCT, alternative lines of therapy should be used.
There are many exciting and potential options for relapsed AL amyloidosis treatment, but the majority lack large comparative studies to help with sequencing. Over 50% of patients have t(11;14), which induces BCL-2 overexpression and provides the opportunity to use a venetoclax-based regimen.22 As of now, though, venetoclax lacks prospective data, but it is still a guideline-recommended regimen. Ixazomib-dexamethasone did not reach its primary endpoint of hematologic overall response compared to the physician’s choice, but patients tolerated treatment for a longer duration. Although data is limited to smaller studies pertaining to carfilzomib, there are favorable positive hematologic response rates, and it may be useful in non-cardiac AL amyloidosis or severe neuropathy. Targeting CD38 with daratumumab or isatuximab also has potential if the patient has a prolonged response to initial treatment with a CD38 targeting agent or if the patient treatment naïve. Utilizing lenalidomide or pomalidomide has also shown a positive response, but not in a phase III trial. Lenalidomide is less well tolerated in AL amyloidosis due to a higher rate of adverse events and increases in BNP and NT-proBNP noted, but starting at a lower dose of 10-15 mg may improve tolerability.5,23
Figure 3: Treatment Management for Relapsed/Refractory AL Amyloidosis


The Future of AL Amyloidosis Treatment
With the need to figure out how to treat AL amyloidosis in the relapsed setting, novel therapies are moving away from suppressing the plasma cell clone to creating therapies that target the removal of existing amyloid deposits. Already deposited amyloid fibrils can continue to cause organ damage progressively, and organ recovery is often delayed in patients who achieve a hematological response.24
Anselamimab (CAEL-101), an anti-amyloid fibril monoclonal antibody, is designed to bind to extracellular amyloid, promote phagocytic clearance, and accelerate organ recovery, particularly in patients with significant, confirmed cardiac involvement. A phase 2 study led by Valent et al. that combined CyBorD and anselamimab in 25 patients across Mayo stage I-IIIa and revealed a robust cardiac response; 50% remained stable, and only 5% demonstrated disease progression.25,26 Unfortunately, anselamimab did not meet the primary endpoints of all-cause mortality and frequency of cardiovascular hospitalization when compared with CyBorD alone in the Phase 3 CARES trial.
Similarly, birtamimab, an amyloid depleter, showed promising results in the phase 2 study that included Mayo stage IV patients, but ultimately did not meet the primary endpoint of all-cause mortality in the phase 3 AFFIRM-AL clinical trial when compared to the standard of care bortezomib-based regimen.
Another promising avenue is AT-02, an IgG1 fusion protein, specifically designed to bind all forms of amyloid and promote phagocytic clearance of existing tissue deposits. AT-02 is specifically designed to remove deposited amyloid, potentially restoring organ function, even in patients in a CHR. In a phase 1/2 study led by Bell et al., AT-02 demonstrated the ability to improve renal function in patients with AL amyloidosis. Patients receiving 2,500 mg every two weeks experienced a mean eGFR increase of 16.3 mL/min/1.73 m2 over 40 weeks, with proteinuria also demonstrating marked reductions. There was no evidence of new or worsening proteinuria, suggesting that organ damage was not only halted, but may also be potentially reversed in select patients.27
B-cell maturation antigen (BCMA)-directed therapies, including CAR-T cells and bispecific T-cell engagers, are promising for eliminating amyloid-producing plasma cells; however, risks such as cytokine release syndrome and neurotoxicity are substantially higher and must be carefully monitored.28 Belantamab mafodotin, a BCMA-directed antibody drug conjugate, has shown a positive response in the relapsed AL amyloidosis setting in a phase II study by Kastritis et al. Mayo cardiac stage I-IIIa and symptomatic organ involvement were included, with 11 patients (31.4%) achieving a VGPR or better; however, ocular toxicities were noted in all but one patient (97.1%) and need to be monitored.29
AL Amyloidosis Conclusion
Light chain amyloidosis is often underdiagnosed due to the detriments from misshapen amyloid fibrils going unnoticed until symptomatic and severe organ damage arises. Compared with treatments from the 1990s to the present, there have been advances in incorporating therapies that reduce amyloid fibril production, even in advanced cardiac and renal stages; however, one main question remains – what should be selected after progression from Dara-CyBorD? In the absence of a standard of care in the relapsed setting, there remains significant room for the development of agents that can successfully dismantle existing fibrils, improve organ function, and ultimately halt the amyloid cascade altogether.
References
-
Baker KR. Light Chain Amyloidosis: Epidemiology, Staging, and Prognostication. Methodist Debakey Cardiovasc J. 2022;18(2):27-35. Published 2022 Mar 14.
-
Al Saleh As, Sidiqi Mh, Muchtar E, Et Al. Outcomes Of Patients With Light Chain Amyloidosis Who Had Autologous Stem Cell Transplantation With 3 Or More Organs Involved. Biology Of Blood And Marrow Transplantation: Journal Of The American Society For Blood And Marrow Transplantation. 2019;25(8):1520-1525.
-
Ashutosh Wechalekar, Alberico Del Torto, Quarta C, Liedtke M. Al Amyloidosis For Cardiologists. Jacc: Cardiooncology. 2022;4(4):427-441.
-
Bell G, Sherman C, Meldorf M, Et Al. Renal Responses In A Phase 1/2 Study Of At-02, A Novel Pan-Amyloid Depleter Ig Fusion Protein For The Treatment Of Patients With Al Amyloidosis. Blood. 2025;146(Supplement 1):693-693.
-
Sanchorawala V. Systemic Light Chain Amyloidosis. N Engl J Med. 2024;390(24):2295-2307
-
Sarubbi C, Abowali H, Varga C, Landau H. Treatment Of Al Amyloidosis In The Era Of Novel Immune And Cellular Therapies. Front Oncol. 2024;14:1425521. Published 2024 Jun 28.
-
Gertz MA, Lacy MQ, Dispenzieri A, et al. Stem cell transplantation compared with melphalan plus dexamethasone in the treatment of immunoglobulin light-chain amyloidosis. Cancer. 2016;122(14):2197-2205.
-
Jaccard A, Moreau P, Leblond V, et al. Autologous Stem Cell Transplantation (ASCT) versus Oral Melphalan and High-Dose Dexamethasone in Patients with AL (Primary) Amyloidosis: Results of the French Multicentric Randomized Trial (MAG and IFM Intergroup). Blood. 2020;106 (11): 421
-
Gertz MA, Dispenzieri A. Systemic Amyloidosis Recognition, Prognosis, and Therapy: A Systematic Review. JAMA. 2020;324(1):79-89.3
-
Nguyen Vp, Landau H, Quillen K, Et Al. Modified High-Dose Melphalan And Autologous Stem Cell Transplantation For Immunoglobulin Light Chain Amyloidosis. Biol Blood Marrow Transplant. 2018;24(9):1823-1827.
-
Kastritis E, Leleu X, Arnulf B, Et Al. Bortezomib, Melphalan, And Dexamethasone For Light-Chain Amyloidosis. J Clin Oncol. 2020;38(28):3252-3260
-
Palladini G, Sachchithanantham S, Milani P, Et Al. A European Collaborative Study Of Cyclophosphamide, Bortezomib, And Dexamethasone In Upfront Treatment Of Systemic Al Amyloidosis. Blood. 2015;126(5):612-615.
-
Venner Cp, Lane T, Foard D, Et Al. , Cyclophosphamide, Bortezomib, And Dexamethasone Therapy In Al Amyloidosis Is Associated With High Clonal Response Rates And Prolonged Progression-Free Survival., Blood, 2012, Vol. 119 19(Pg. 4387-4390)
-
Mikhael Jr, Schuster Sr, Jimenez-Zepeda Vh, Et Al. , Cyclophosphamide-Bortezomib-Dexamethasone (Cybord) Produces Rapid And Complete Hematologic Response In Patients With Al Amyloidosis., Blood, 2012, Vol. 119 19(Pg. 4391-4394)
-
Kastritis E, Palladini G, Minnema Mc, Et Al. Daratumumab-Based Treatment For Immunoglobulin Light-Chain Amyloidosis. N Engl J Med. 2021;385(1):46-58.
-
Oubari S, Hegenbart U, Schoder R, Et Al. Daratumumab In First-Line Treatment Of Patients With Light Chain Amyloidosis And Mayo Stage Iiib Improves Treatment Response And Overall Survival. Haematologica. Published Online July 13, 2023.
-
Yohannan B, Rees M, Gertz Ma, Et Al. Improved Survival With Daratumumab-Cybord Compared With Cybord As Frontline Therapy For Al Amyloidosis. Blood Neoplasia. 2025;2(2):100092. Published 2025 Mar 10.
-
Kastritis E, Minnema Mc, Dimopoulos Ma, Et Al. P915: Efficacy And Safety Of Daratumumab Monotherapy In Newly Diagnosed Patients With Stage 3b Light Chain Amyloidosis: A Phase 2 Study By The European Myeloma Network. Hemasphere. 2022;6(Suppl ):805-806. Published 2022 Jun 23.
-
Varga C. Autologous Stem Cell Transplantation in Light Chain Amyloidosis: The Ultimate Treatment?. Transplant Cell Ther. 2022;28(2):57-58.
-
Romera I, Verdejo J, Inés Viviente, Et Al. Autologous Hematopoietic Cell Transplantation In Al Amyloidosis: A Single-Center Experience. Blood. 2024;144(Supplement 1):6871-6871.
-
D'Souza A, Dispenzieri A, Wirk B, et al. Improved Outcomes After Autologous Hematopoietic Cell Transplantation for Light Chain Amyloidosis: A Center for International Blood and Marrow Transplant Research Study. J Clin Oncol. 2015;33(32):3741-3749.
-
Alvanidis G, Kotsos D, Frouzaki C, Fola A, Hatjiharissi E. The potential role of BCL-2 inhibition in amyloidosis and plasma cell leukemia. Front Oncol. 2025;15:1549891. Published 2025 Mar 21.
-
Wechalekar Ad, Cibeira Mt, Gibbs Sd, Et Al. Guidelines For Non-Transplant Chemotherapy For Treatment Of Systemic Al Amyloidosis: Eha-Isa Working Group. Amyloid. 2023;30(1):3-17.
-
Wang J, Li J, Zhong L. Current Status And Prospect Of Anti-Amyloid Fibril Therapy In Al Amyloidosis. Blood Reviews. 2024;66:101207.
-
Valent J, Silowsky J, Kurman Mr, Et Al. Cael-101 Is Well-Tolerated In Al Amyloidosis Patients Receiving Concomitant Cyclophosphamide-Bortezomib-Dexamethasone (Cybord): A Phase 2 Dose-Finding Study (Nct04304144). Blood. 2020;136(Supplement 1):26-27.
-
Valent J, Liedtke M, Zonder J, Et Al. S204: Safety And Tolerability Of Cael-101, An Anti-Amyloid Monoclonal Antibody, Combined With Anti-Plasma Cell Dyscrasia Therapy In Patients With Light-Chain Amyloidosis: 18-Month Results Of A Phase 2 Study. Hemasphere. 2023;7(S3):E2801725-E2801725.
-
Bell G, Sherman C, Moldorf M Et Al. Renal Responses In A Phase 1/2 Study Of At-02, A Novel Pan-Amyloid Depleter Ig Fusion Protein For The Treatment Of Patients With Al Amyloidosis. Blood. Abstract 693 (2025).
-
Kfir-Erenfeld S, Asherie N, Grisariu S, Avni B, Zimran E, Assayag M, Et Al. Feasibility Of A Novel Academic Bcma-Cart (Hbi0101) For The Treatment Of Relapsed And Refractory Al Amyloidosis. Clin Cancer Res. (2022) 28:5156–66.
-
Kastritis E, Palladini G, Dimopoulos Ma, Et Al. S198: Efficacy And Safety Of Belantamab Mafodotin Monotherapy In Patients With Relapsed Or Refractory Light Chain Amyloidosis: A Phase 2 Study By The European Myeloma Network. Hemasphere. 2023;7(Suppl ):E2301416. Published 2023 Aug 8.